
Proverka Solutions Private Limited is a leading specialist provider of validation services. We have
grown as a reputable company and offer our services nationally & internationally.
The pharmaceutical industry is a highly regulated environment based on research, evidence, record-
keeping and validation. Our experience and expertise will give you a clear, compliant and
transparent validation package.
The term "Thermal validation" is the process of validating / qualifying equipment and storage
facilities to prove that they will create and maintain the temperatures they are designed for.
Our range of bespoke services to the pharmaceutical, biotechnology and healthcare industries.
Validation and Monitoring equipment (Wired & Wireless) are major responsibilities comes under
thermal validation. Well maintained and calibrated equipment ensures that it operates at peak
performance and complies with regulatory requirements.
We are specialises in the qualification of the following equipment:
Autoclave / Sterilizer Qualification • Cold rooms, Warehouse and Room Mapping • Depyrogenation Ovens Qualification • Lyophilizers Qualification • Depyrogenation Tunnels • BOD, Deep Freezers / Walk in Freezers • Photostability Chamber Qualification • Steam-in-Place Qualification • Incubators • Stability Cabinets • Washer Disinfectors • CIP Systems • Dry Heat Ovens / Hot Air oven • Furnaces • SIP Systems • Ultra Low Freezers
Autoclave / Sterilizer Qualification




Autoclave Validation / Qualification is mandatory for all machines used for biological sterilization, in the biomedical and pharmaceutical industries within the FDA, WHO & EU controlled areas. Sterilization can be accomplished by either physical or chemical means. This benchtop autoclave validation sterilizer. As used for sterilization of instruments and porous loads in hospitals, private clinics and dental and veterinary offices. The principal physical means is autoclaving; other physical methods include boiling and dry heat. Chemicals used for sterilization include the gases ethylene oxide and formaldehyde and liquids such as glutaraldehyde. Of all these sterilants, autoclaving is the fastest, most reliable and hence; most commonly used within the FDA, WHO and EU zones of influence. It must always be remembered that it is also one of the easiest processes to get wrong. This is why regulators will nearly always scrutinize and ask about validation methods used in autoclave qualification / Qualification activities. Autoclave validation / Qualification must follow the routine validation / Qualification document string of; VP – URS – DQ – VRA – IQ – OQ – PQ
Cold Rooms, Warehouse and Room Mapping



Cold Chain Validation suddenly became a hot topic when the results of a government investigation into Cold Chain distribution compliance were published. The facts release indicated that nearly a quarter (23&precnt;) of all drugs distributed in the USA, are of unknown efficacy. The distribution of these temperature sensitive products in vehicle containers that lacked validated methods of maintaining the correct internal temperature or even the ability to produce a validated history of what the actual conditions were; has rendered these products 'adultered', i.e., of unknown efficacy or safety. Vehicle body internal temperatures; with seasonal variations, can range from a high of 50 to a low of -40 (degrees centigrade). In contrast the mandated internal temperature for distribution containers for standard drugs and medical products is 2 to 8 (degrees centigrade), with much lower temperatures specified for some drugs. This shocking news has certainly guaranteed that Cold Chain Validation compliance will be top of everyone's audit list, specifically when drug interactions with the end user are considered as critical. The implications of products being damaged and out of specification may induce many companies to replace doubtful goods or even trigger the regulators into initiating product recalls. This problem is expected to create a veritable stampede as distributors scramble to ensure that all the vehicles and containers they used in this essential everyday service, are rendered Cold Chain compliant, as soon as is possible.
Depyrogenation Ovens Qualification

Dry heat sterilization (or Depyrogenation) is a process aimed at the reduction in the level of pyrogens with the use of hot air in temperature ranging from 160° C up to 400° C. The temperature used depends on the duration of the process. Gravity or mechanical heat convection can be used for this process. The former uses the natural interaction between air and different temperatures and the latter produces a specific flow of air with the help of a blower. Depyrogenation is mainly used in the sterilization of vials for aseptic filling. The process is also useful to sterilize assembled and packaged materials, since heat conduction does not require the contact of the product with steam or water. However, not all products can withstand the high temperatures required for this kind of process (e.g. metal goods, heat sensitive solutions, etc.). Batch and dynamic are the two main techniques for Depyrogenation. Batch production is usually done in static ovens where the product is placed in metal crates and exposed to high temperatures. Dynamic involves the use of a Depyrogenation tunnel, where goods are transported over a continuous band through different heating zones of the tunnel.
Lyophilizers Qualification

Freeze drying (or lyophilization) is a technique used to remove water from a solution to leave a dry ‘cake’ as an end product. The dry product is much more stable than a solution. The reduced weight and volume is a big advantage and the removal of the water does not change the activity of the product. This technique involves freezing the product on a shelf using extremely low temperatures (typically –40 to –50°C), applying a vacuum and then slowly heating the product to ensure the ice turns into vapor. This removes the water content from product by a process called sublimation. The critical parameters during freeze-drying are temperature (product and shelf) and pressure/Vacuum. These measurements are usually recorded with every cycle to determine when the product is dry. In addition to monitoring each cycle, freeze driers also require regular validation. This will generally involve temperature mapping of the shelves in the chamber, condenser cool down test, system evaluation test (Vacuum Pull-Down) to test Vacuum pumping system efficiency and Vacuum integrity test to show quality of seals, as well as validation of steam supplies for the SIP system. As freeze drying is a rather expensive process most products being processed are within the pharmaceutical and biotechnology industries. Companies often use freeze-drying to increase the shelf life of products, such as vaccines and other injectables. By removing the water from the material and sealing the material in a vial, the material can be easily stored, shipped and later reconstituted to its original form for injection. Lyophilization can also be performed in other industries like: Food industry (example: freeze dried fruits, astronaut food supplies, instant coffee) Technological industry: • In chemical synthesis to make them more stable, or easier to dissolve in water for subsequent use • In bio separations as a late-stage purification procedure to remove solvents In bacteriology to conserve special strains Conservation and preservation industry: • Flowers • Animals Water damaged books and documents
Depyrogenation Tunnels
Pasteurization Tunnels
BOD, Deep Freezers / Walk in Freezers

A freezer is a storage unit used in pharmaceutical industry to store contents such as samples and other medical material just below the freezing temperature. There are basically two styles of freestanding freezers - chest and upright models. Freezers have a long lifespan with most models having an expected lifecycle of twenty years for chest models and shorter for uprights, especially self-defrost models. There are also walk-in style freezers for bulk storage.
Photostability Chamber Qualification

Environmental chamber Environmental test chambers feature precise control of appropriate parameters to create repeatable environmental conditions. Test chambers are suitable for numerous applications: product shelf life, stability and package testing, light exposure and temperature evaluation studies, electronic component burn-in, plant growth and many more. Environmental testing chambers come in various designs and sizes, ranging from a few liters (chamber) up to several thousand cubic meters (room) and feature a wide temperature and relative humidity (RH) range. Most modern test chambers offer microprocessor-based controllers with computer communications, autotune, solid state RH sensor and stainless-steel temperature probes. LED displays show both process and set point values. Tests are performed according to the HALT or HASS testing principles: • HALT - Highly Accelerated Life Test • HASS - Highly Accelerated Stress Screen HALT and HASS, also known as Accelerated Stress Testing (AST), subject a product to a series of overstresses, effectively forcing product weak links to emerge by accelerating fatigue. In other words, tests are unique. Accelerated product reliability test methods focus on finding defects in products so they can be fixed before becoming expensive field issues. Stresses are applied in a controlled, incremental fashion while the unit under test is continuously monitored for failures. Once the weaknesses of the product are detected the limits of the product are clearly defined.
Steam-in-Place Qualification

Steam-in-place (SIP) cleaning uses the thermal energy of condensing steam for a controlled time to clean and disinfect the interior surfaces of bioreactors, vessels, tanks, piping, filters and associated fittings "in place," without costly removal of the target equipment, or time-consuming disassembly of critical production lines. High-pressure steam is repeatedly introduced into the product-contacting areas of the processing system, for sufficient duration required to reduce bioburden and neutralize contaminants. The success of the steam cleaning is validated by thermal mapping, where temperature sensors are placed throughout the system before steam-in-place cleaning to ensure that specified temperatures and exposure times are achieved. The temperature sensors or thermocouples are typically placed throughout the system via flanges and gaskets, eliminating the need to disassemble the system. With years of experience in steam-in-place validation, Masy Bio Services can help ensure the sterility of your production facility and the corresponding safety and efficacy of your products. A pre- execution review of your production line design and evaluation of your test plan requirements will help us identify the critical areas and "problem spots" of your system, helping to ensure a successful validation of your steam-in-place sterilization.
Incubators

In microbiology an incubator is a device for controlling temperature, humidity and other conditions under which a microbiological culture is being grown. The simplest incubators are insulated boxes with an adjustable heater, typically going up to approx. 65°C. Some incubators can go slightly higher but generally to no more than 100°C. More advanced incubators may have the ability to lower the temperature (via refrigeration) and/or the ability to control humidity or CO2 levels. This feature is specifically designed for cell growth. Most incubators provide inner and outer glass doors for transparency. The main controller includes a timer. Some can also be programmed to cycle through different temperatures, humidity levels, etc. Incubators can vary in size from tabletops units to units the size of small rooms. Incubators may also provide special features such as regulation of shake speed. As for temperature, 37°C is a commonly used temperature in incubators being the normal human body temperature. Most bacteria, especially the frequently used E. Coli, grows well under such conditions. For other applications lower temperatures are required, (for fish cells a growth temperature of 25°C is optimal).
Washer Disinfectors

Washer/disinfectors are used in medical, as well as pharmaceutical applications, to either prepare goods for future autoclaving (for example surgery instruments to remove residues such as blood and protein) or to disinfect (semi sterilize) goods for later use. In the case of steam sterilization, the surface has to be properly cleaned. Without proper cleaning, sterilization is not possible. A clean and debris-free surface is required during steam sterilization allowing steam to contact all instrument surfaces. It has been proven that bacteria can survive a steam sterilization cycle if they are protected under dried debris. When the washer/disinfection cycle is complete, the instruments can be inspected, placed in sterilization pouches or wraps and then properly sealed and processed in the sterilizer.
CIP Systems
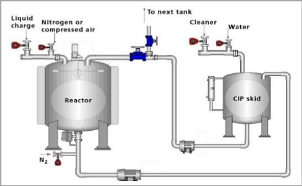
Validation of cleaning procedures has generated considerable discussion since agency documents, including the Inspection Guide for Bulk Pharmaceutical Chemicals and the Biotechnology Inspection Guide, have briefly addressed this issue. These Agency documents clearly establish the expectation that cleaning procedures (processes) be validated. This guide is designed to establish inspection consistency and uniformity by discussing practices that have been found acceptable (or unacceptable). Simultaneously, one must recognize that for cleaning validation, as with validation of other processes, there may be more than one way to validate a process. In the end, the test of any validation process is whether scientific data shows that the system consistently does as expected and produces a result that consistently meets predetermined specifications.
Dry Heat Ovens / Hot Air Oven

Components and products. Dry heat sterilization is often used for heat-stable oils, ointments and powders. Most often, Depyrogenation of parenteral containers is performed utilizing a dry heat oven. The Depyrogenation process is also utilized on certain heat-stabile components, glass containers, metal equipment, etc. to render the item and final parenteral product free of pyrogens. The equipment utilized to provide the dry heat medium must be validated to ensure that the system is able to provide sterile and/or dehydrogenated components, on a reproducible basis. The validation of a dry heat sterilization and Depyrogenation process involves approaches and procedures which parallel those utilized for steam sterilization. The efficiency of any heat treatment is determined by the design and source of the heat. Hot air is substantially less efficient in a thermal transfer medium as compared to steam. The validation effort must include heat distribution, heat penetration, bioburden and pyro burden determination, filter integrity and microbial/endotoxin challenges.
Furnaces

Temperature Uniformity Surveys commonly referred to as a TUS, establish objective evidence of the heat distribution characteristics throughout an oven or furnace system. It also identifies the qualified work zone within the oven or furnace system. TUS's can also be an effective way of determining the efficiency of the furnace system and to monitor furnace performance over time. System Accuracy Test, commonly referred to as an SAT, is another critically important and often required test. System Accuracy Tests are performed to determine the error within the temperature control system (temperature control device/lead wire/thermocouple). Regardless of the application, knowing the error that exists in your temperature control system allows for more consistent control of quality.
SIP Systems

Stability Chamber
A Sterilization in Place (SIP) installation will usually comprise one or more pieces of processing
equipment, such as a fermenter and a centrifugal separator to handle harvests, connected by rigid
stainless steel or flexible Teflon®-lined piping. The installation will be capable of withstanding steam
pressure up to, say, 20 psi and corresponding sterilizing temperatures in the 121° to 125°C range.
There will be a supply of steam suitable for the procedure, under pressure control and a "trapped"
drain at a low point on the system, which will pass condensed water, but not steam. The safe
operation of the installation will be controlled by suitable safety valves or "burst disks".
Design qualification of a SIP installation will require confirmation that the process equipment, pipe
work and steam supply equipment meet preset specifications for materials and for pressure and
temperature resistance. Attention must be paid to the quality of the steam, which will be used. This
usually means that the steam is generated in a dedicated "clean-steam" generator. The steam may
also pass a micro-filter before use. The cleanliness of the steam must be maintained by the use of
pressure-grade stainless steel or Teflon®-lined tubing and suitably constructed pressure control and
shut-off valves and pressure gauges.
Other design aspects of the equipment intended for SIP will include ensuring that the steam can
reach all parts of the equipment in contact with product and that air is not trapped in the plant during
sterilization. There must also be a means for the easy clearance of condensate during the heating
process, through the steam trap. Finally, temperature sensors must be sited where they will
represent reliably the state of the equipment during the sterilization cycle. Often, the most favored
point for temperature measurement is at the condensate drain, since this will be the last area to
reach operating temperature. However, there may be good reasons for siting thermo-sensors in
difficult-to-reach areas of the plant.
Installation qualification of a well-designed SIP system will involve confirming the proper installation
of the process equipment and correct siting and connections for all pipe work, including ensuring that
proper condensate drainage can occur. All required services and monitoring devices should be in
place. Operational qualification starts with the start-up of the steam generation set-up and
confirmation that correct pressure and steam volume is achieved. The sterilizing cycle is then run.
Steam under pressure is passed through the entire installation while allowing the escape of air
through properly placed vents in the piping or on the equipment. These vents are usually protected
by steam-resistant bacterial filters. After a suitable period of steaming, the air vents are closed and
steam pressure is allowed to build to the required level. Pressure is maintained during a preset
period, then the steam is released through a condenser. Temperature sensors in the system should
indicate that the recorded pressure resulted in the required temperature being reached for sufficient
time to ensure destruction of all contaminants. Cooling down of the equipment requires that air be
allowed to pass back into the system through one or more of the sterilizing filters on the vents, to
prevent the development of a vacuum.
Validation of the system operations will require the use of chemical and biological indicators, which
should be placed in sections of the plant determined to be difficult to free of air and near the
condensate drain. The prescribed sterilization cycle should always yield sterile indicators. The
process can be challenged by "worst-case" conditions, such as a reduction in sterilization period or
steam pressure. Eventually, a set of operating conditions, which have been shown to produce sterile
plant reproducibly, can be adopted as the validated process. Minimum and maximum limits on the
steam pressure, indicated temperature and exposure time will be set. If appropriate, the final
evidence of the validity of the sterilization process may be achieved by running culture medium
through the sterilized set up in imitation of the manufacturing processes and then incubating a large
number of media samples, or the entire batch, to ensure no bacterial growth occurs. In any event,
the regular manufacture of product batches, which pass sterility and endotoxin tests after processing
in the SIP plant, will confirm PQ of the system. These records, however, will not substitute for proper
SIP process validation.
Stability Chamber

Stability chambers are ideal for testing storage, shelf life, packaging and stability. Stability chambers are available in temperature only or with temperature and humidity. Sizes range from reach-in to walk-in and most chambers feature a temperature range of -20° C to 60° C and an optional humidity range from 10% to 95% RH. Stress testing is likely to be carried out on a single batch of the drug substance. It should include the effect of temperatures in 10° C increments (50° C, 60° C, etc.). Increments may be greater for accelerated testing. Stress testing for humidity typically is performed at 75% RH or greater (where appropriate). Other tests can include effects of oxidation and photolysis. Stress testing of the drug substance can help identify the likely degradation products, which can in turn help establish the degradation pathways and the intrinsic stability of the molecule. It can also validate the stability indicating power of the analytical procedures used. The nature of the stress testing will depend on the individual drug substance and the type of drug product involved.


